I recently helped put out a preprint on combining active learning with docking for virtual screening applications.
Abstract
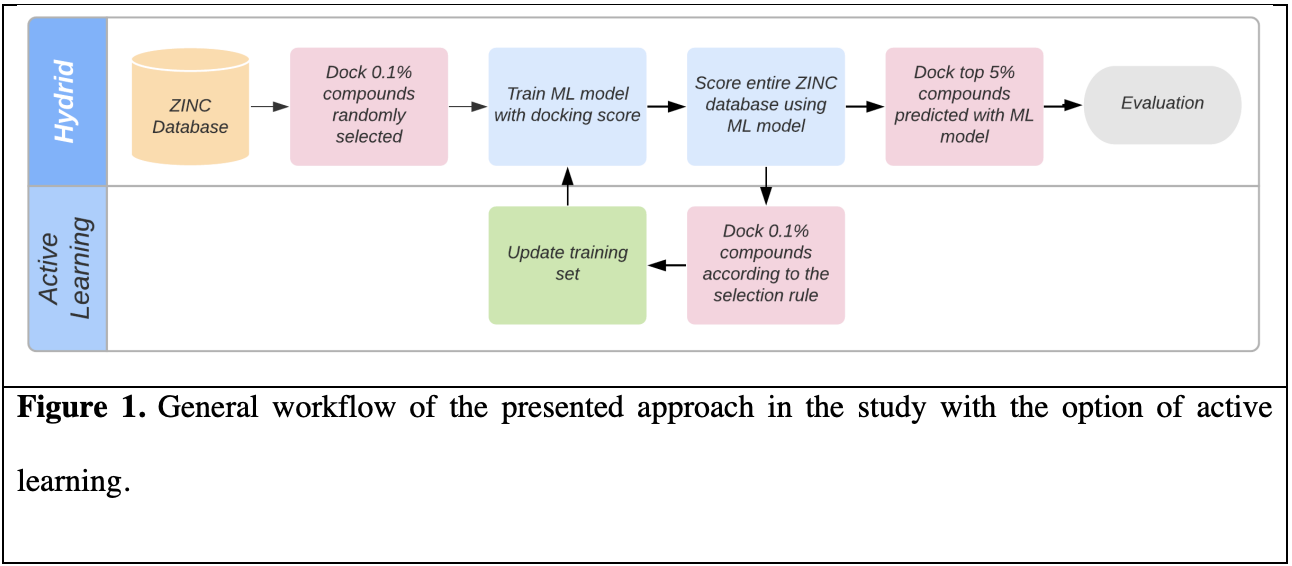
With the advent of make-on-demand commercial libraries, the number of purchasable compounds available for virtual screening and assay has grown explosively in recent years, with several libraries eclipsing one billion compounds. Today’s screening libraries are larger and more diverse, enabling discovery of more potent hit compounds and unlocking new areas of chemical space, represented by new core scaffolds. Applying physics-based in-silico screening methods in an exhaustive manner, where every molecule in the library must be enumerated and evaluated independently, is increasingly cost-prohibitive. Here, we introduce a protocol for machine learning-enhanced molecular docking based on active learning to dramatically increase throughput over traditional docking. We leverage a novel selection protocol that strikes a balance between two objectives: (1) Identifying the best scoring compounds and (2) exploring a large region of chemical space, demonstrating superior performance compared to a purely greedy approach. Together with automated redocking of the top compounds, this method captures nearly all the high scoring scaffolds in the library found by exhaustive docking. This protocol is applied to our recent virtual screening campaigns against the D4 and AMPC targets that produced dozens of highly potent, novel inhibitors, and a blinded test against the MT1 target. Our protocol recovers more than 80% of the experimentally confirmed hits with a 14-fold reduction in compute cost, and more than 90% of the hit scaffolds in the top 5% of model predictions, preserving the diversity of the experimentally confirmed hit compounds.
Check out the preprint here .