Abstract
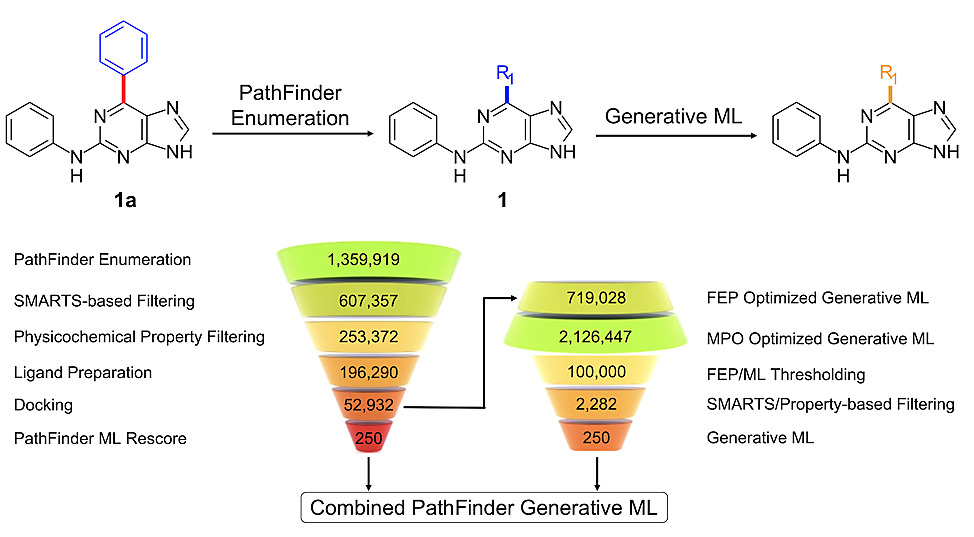
The hit identification process usually involves the profiling of millions to more recently billions of compounds either via traditional experimental high throughput screens (HTS) or computational virtual high throughput screens (vHTS). We have previously demonstrated that by coupling reaction-based enumeration, active learning and free energy calculations, a similarly large-scale exploration of chemical space can be extended to the hit-to-lead process. In this work, we augment that approach by coupling large scale enumeration and cloud-based FEP profiling with goal-directed generative machine learning, which results in a higher enrichment of potent ideas compared to large scale enumeration alone, while simultaneously staying within the bounds of a predefined drug-like property space. We are able to achieve this by building the molecular distribution for generative machine learning from the PathFinder rules-based enumeration and optimizing for a weighted sum QSAR based multi-parameter optimization function. We examine the utility of this combined approach by designing potent inhibitors of cyclin-dependent kinase 2 (CDK2) and demonstrate a coupled workflow that can: (1) provide a 6.4 fold enrichment improvement in identifying < 10nM compounds over random selection, and a 1.5 fold enrichment in identifying < 10nM compounds over our previous method (2) rapidly explore relevant chemical space outside the bounds of commercial reagents, (3) use generative ML approaches to “learn” the SAR from large scale in silico enumerations and generate novel idea molecules for a flexible receptor site that are both potent and within relevant physicochemical space and (4) produce over 3,000,000 idea molecules and run 2153 FEP simulations, identifying 69 ideas with a predicted IC50 < 10nM and 358 ideas with a predicted IC50 <100 nM. The reported data suggest combining both reaction-based and generative machine learning for ideation results in a higher enrichment of potent compounds over previously described approaches, and can rapidly accelerate the discovery of novel chemical matter within a predefined potency and property space.
Table 1
Cumulative Predicted potencies of Pathfinder Random, Pathfinder Rescore, Generative ML and Pathfinder Generative ML Designs.
| Prioritization Approach | #Profiled | <10nM | <100nM | <1µM | >1µM |
|---|---|---|---|---|---|
| PathFinder Random | 935 | 9 (1.0%) | 56 (5.9%) | 205 (22%) | 730 (77%) |
| PathFinder Rescore | 500 | 21 (4.2%) | 120 (24%) | 322 (64%) | 178 (36%) |
| Generative ML | 500 | 31 (6.2%) | 139 (28%) | 314 (63%) | 186 (37%) |
| PathFinder Generative ML | 500 | 32 (6.4%) | 145 (29%) | 341 (68%) | 159 (32%) |