Abstract
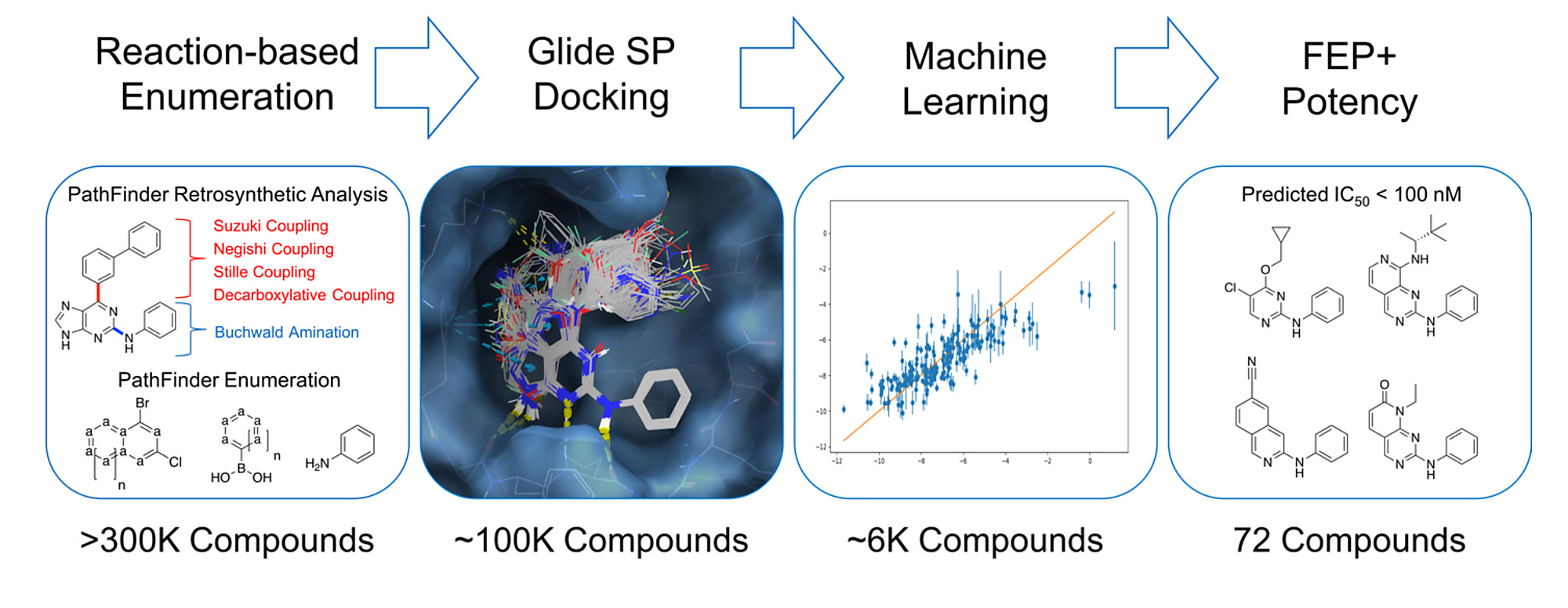
The hit-to-lead and lead optimization processes usually involve the design, synthesis, and profiling of thousands of analogs prior to clinical candidate nomination. A hit finding campaign may begin with a virtual screen that explores millions of compounds, if not more. However, this scale of computational profiling is not frequently performed in the hit-to-lead or lead optimization phases of drug discovery. This is likely due to the lack of appropriate computational tools to generate synthetically tractable lead-like compounds in silico, and a lack of computational methods to accurately profile compounds prospectively on a large scale. Recent advances in computational power and methods provide the ability to profile much larger libraries of ligands than previously possible. Herein, we report a new computational technique, referred to as “PathFinder”, that uses retrosynthetic analysis followed by combinatorial synthesis to generate novel compounds in synthetically accessible chemical space. In this work, the integration of PathFinder-driven compound generation, cloud-based FEP simulations, and active learning are used to rapidly optimize R-groups, and generate new cores for inhibitors of cyclin-dependent kinase 2 (CDK2). Using this approach, we explored >300 000 ideas, performed >5000 FEP simulations, and identified >100 ligands with a predicted IC50 < 100 nM, including four unique cores. To our knowledge, this is the largest set of FEP calculations disclosed in the literature to date. The rapid turnaround time, and scale of chemical exploration, suggests that this is a useful approach to accelerate the discovery of novel chemical matter in drug discovery campaigns.