Check out a new paper I worked on combining synthetic enumeration, free energy perturbation calculations and active learning.
Abstract
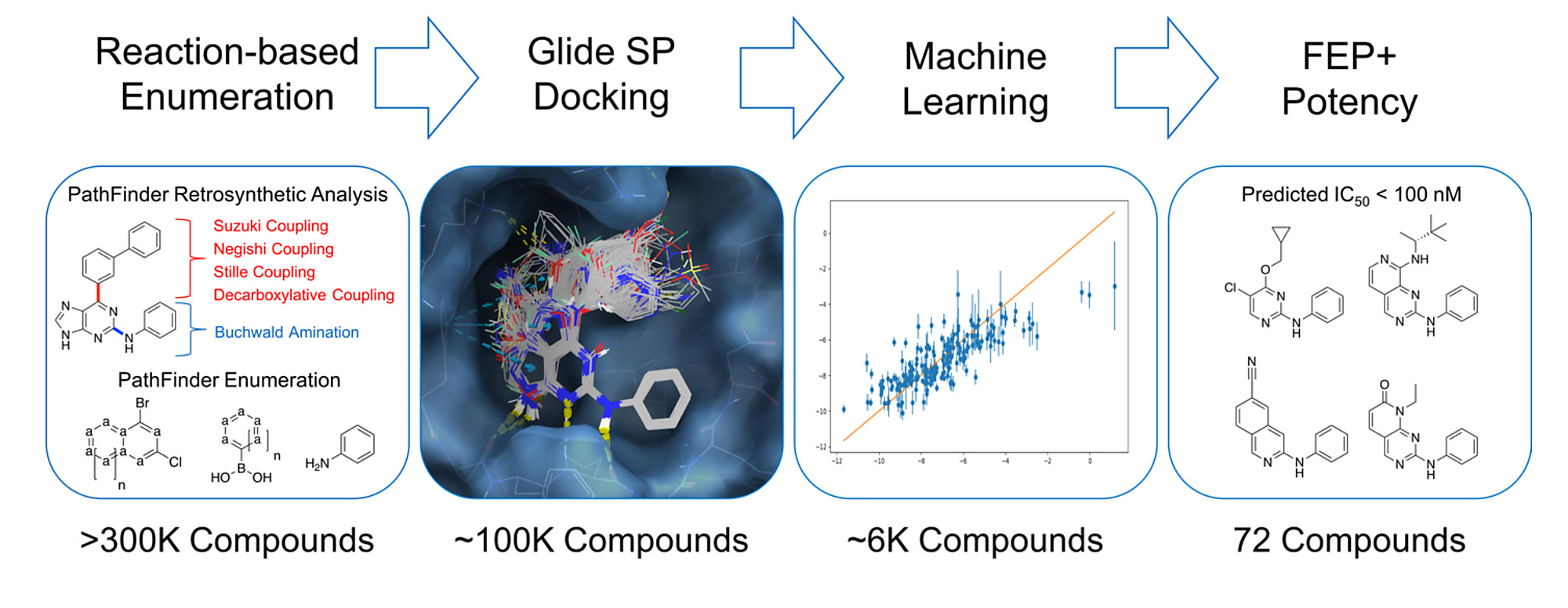
We report a new computational technique, PathFinder, that uses retrosynthetic analysis followed by combinatorial synthesis to generate novel compounds in synthetically accessible chemical space. Coupling PathFinder with active learning and cloud-based free energy calculations allows for large-scale potency predictions of compounds on a timescale that impacts drug discovery. The process is further accelerated by using a combination of population-based statistics and active learning techniques. Using this approach, we rapidly optimized R-groups and core hops for inhibitors of cyclin-dependent kinase 2. We explored greater than 300 thousand ideas and identified 35 ligands with diverse commercially available R-groups and a predicted IC50 < 100 nM, and four unique cores with a predicted IC50 < 100 nM. The rapid turnaround time, and scale of chemical exploration, suggests that this is a useful approach to accelerate the discovery of novel chemical matter in drug discovery campaigns.